Characterising somatic structural variation in colorectal cancer with long nanopore reads
- shared.published_on: May 25 2023
In oncology, the goal of precision medicine is to identify genetic vulnerabilities in a cancer sample and match them to the treatment most likely to be effective. Whilst there has been some clinical success on this front already, there is still considerable basic research to be done to detect the full range of possible variants, determine their functional impact, and use that information to guide the development of new treatments.
The scientific community is making significant strides toward the goal of identifying and classifying cancer-associated variants. However, until recently, most of this research has been performed using short-read sequencing technology, which is largely limited to detecting single nucleotide variants and small insertions and deletions. Structural variants (SVs), which are 50 bp or larger and can be more complex, can easily escape detection in short-read data due to ambiguities in read alignment. By contrast, long nanopore reads can capture full SVs; when read alignment is performed, these SVs are represented accurately and completely.
In a new study, scientists based at Huazhong University of Science and Technology, China, used the Oxford Nanopore PromethION device to perform the first nanopore sequencing analysis of SVs in human colorectal cancer samples1. Their results not only point to the presence of untapped, previously hidden genomic information from SVs, but also suggest that long nanopore sequencing reads can be an effective method for detecting novel gene fusions. Both SVs and gene fusions are known as cancer drivers.
For this project, Xu et al. used nanopore sequencing on 21 pairs of stage II or stage III colorectal tumour clinical research samples and matched normal samples. They also analysed the samples with short-read whole-exome sequencing and short-read RNA sequencing.
Using nanopore sequencing, the longest read generated was nearly 900 kb and the N50 length was almost 43 kb. Sequencing data alignment and SV calling were performed with freely available tools including NGMLR2 and Sniffles2. Each sample contained more than 19,000 SVs, on average, and the team eliminated germline variants to focus on the 500 or so somatic variants in each sample. The authors noted that this is roughly double the number of SVs typically found in short-read studies of cancer samples. In total, they found approximately 5,200 unique somatic SVs.
‘Our results show long-read sequencing precisely and reliably detects 494 somatic SVs per sample’
Xu, L. et al. PLoS Genet. (2023)
The somatic SVs included deletions, insertions, duplications, inversions, and translocations. Interestingly, almost two-thirds of the variants were found in at least two samples, suggesting that there may be common cancer-associated SVs that could represent future druggable targets for colorectal cancer. In addition, the variants occurred in locations that could indicate their role in driving cancer. The authors suggested that ‘some loci with high frequency were associated with the genes involved in oncogenesis and development of [colorectal cancer], including alternative splicing factor RBFOX1, tumor suppressor gene FHIT, and several oncogenes such as LGR6, CTGF and RAB11A'.
In the study, the team pinpointed certain novel SVs for further analysis. For example, they detected a 4.9 Mb inversion on chromosome 5 that covered exon 1 of the APC gene (Figure 1), confirming its presence with Sanger sequencing of the breakpoints. While RNA sequencing data indicated significantly lower APC expression as a result of this change, the short-read exome sequencing data identified no variants in the APC gene, demonstrating the necessity of long nanopore sequencing reads for the identification of such large-scale variants.
Another example comprised an 11.2 kb inversion on chromosome 7 that involved exon 11 of the CFTR gene. This was another high-confidence call, with four reads spanning the full inversion and its breakpoints.
‘The inversions in APC and CFTR clearly altered the structure (including coding regions) of both genes, but were not detected by [whole exome sequencing]’, the authors wrote. The CFTR example, they added, ‘showed that the enhanced read length enables a full capture of SVs, significantly improving cancer SV detection efficacy’.
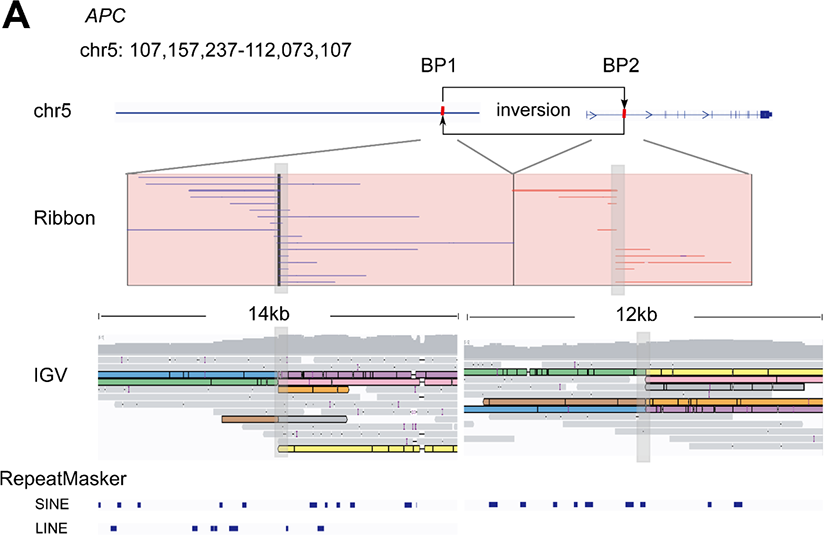
Figure 1: Reads and structures of the 4,915 kbp inversion spanning exon 1 of APC. Figure from Xu, L. et al.1 and available under Creative Commons license (https://creativecommons.org/licenses/by/4.0/).
Beyond SVs, long nanopore reads also allowed the team to identify gene fusions that could have evaded detection with other techniques. Fusions caused by rearrangements in the genome ‘represent an important part of tumor genomic landscape and are involved in development of approximately 16% of all cancer types, including [colorectal cancer]’, Xu et al. noted.
Despite their importance, gene fusions are not always easy to find. Short-read exome or genome sequencing can miss these large elements because reads simply aren’t long enough to capture them, while RNA-Seq based on short reads ‘suffers from poor sensitivity for detecting the fusion genes that are expressed at rather low levels or diluted by accompanying non-cancerous cells’, the scientists wrote. ‘In contrast, the advantages of long-read sequencing allow more effective identification of novel genetic rearrangements that may result in gene fusions’.
In this study, the team cited two examples of gene fusions that were detected using long nanopore reads. The first is RNF38-RAD51B, which was also found with RNA sequencing and confirmed with PCR products of the breakpoint junctions. This fusion is expected to alter the function of RNF38, a gene linked to key driver mechanisms of cancer. The other example is SMAD3-SHISA6, which was also validated with PCR but was not detected in RNA-Seq data. The fusion could cause dysfunction of SMAD3, a transcription factor associated with tumour suppression.
‘Our work highlights the potential of the long-read sequencing in serving as a new platform for the precise diagnosis and treatment of [colorectal cancer]’
Xu, L. et al. PLoS Genet. (2023)
Noting that these gene fusions require additional study, the authors reported that their results ‘suggest that nanopore sequencing may serve as a new strategy for detecting oncogenic gene fusions’.
Xu, L. et al. PLoS Genet. 19(2):e1010514 (2023).
2. Sedlazeck, F.J. Nat. Methods 15(6):461-468 (2018).






