Nanopore sequencing improves surveillance of serious respiratory diseases and characterises the causative agents
)
- shared.published_on: November 8 2023
Respiratory tract infections are a leading cause of illness and death — in particular, lower respiratory infections (LRTI), which accounted for infections in over 488 million people and 2.4 million deaths in 20191. Respiratory infections are caused by a variety of infectious agents — bacterial, viral, and fungal — but identifying which agent is the cause is not always feasible. In China, there are approximately 10 million new cases of LRTIs each year, with a 30% mortality rate, but the cause of 30% of total LRTI cases is unknown2. This is due to the limitations of successfully culturing and identifying microorganisms using traditional methods, such as PCR and serological techniques2.
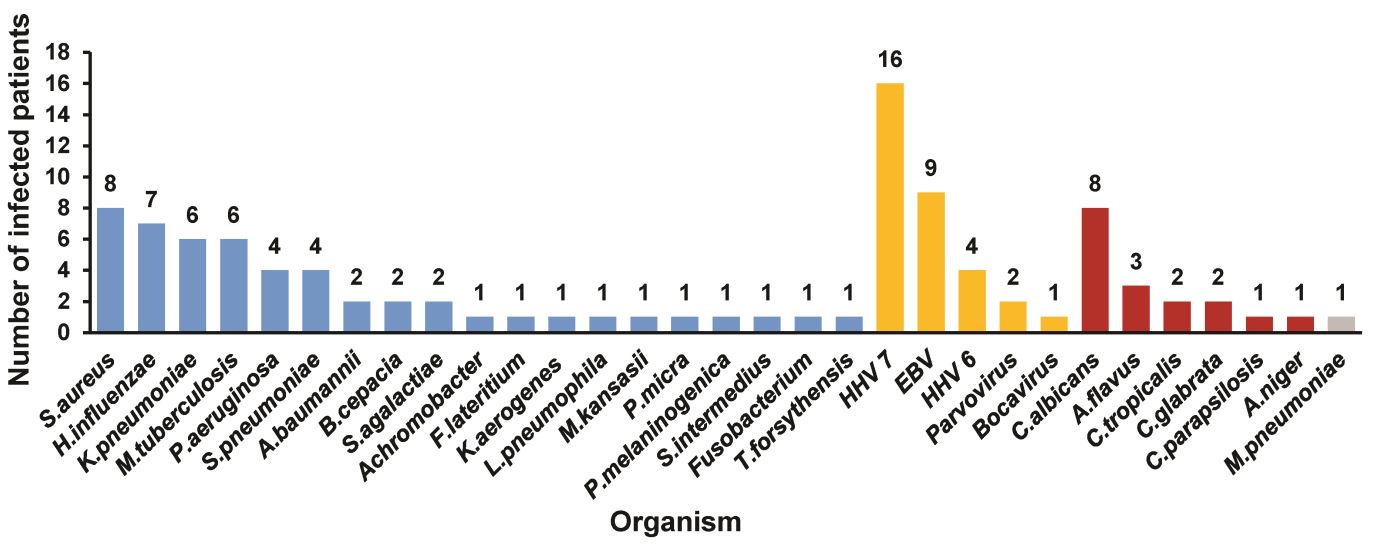
Using a metagenomic approach, Luo et al.tested the potential of nanopore sequencing to identify LRTI infections2. They performed nanopore sequencing of 70 bronchoalveolar lavage fluid research samples, and compared its performance to traditional culture methods and qPCR. Libraries were prepared using the Rapid PCR Barcoding Kit and sequenced on a MinION; real-time pathogen identification was then performed during sequencing. Using nanopore sequencing, pathogens were identified in 59 samples; qPCR identified pathogens in 56 samples, while culture methods found pathogens in just 18. In this study, nanopore sequencing outperformed traditional culture methods and, in addition, could also identify antimicrobial resistance genes and viruses. Moreover, the turnaround time for the nanopore sequencing method was six hours, compared to three days using traditional methods, demonstrating the future potential for this approach to rapidly identify pathogens.
Legionnaire’s disease is a serious respiratory infection caused by Legionella spp. bacteria, and can result in severe pneumonia and death. In an effort to characterise the diverse strains of this single respiratory disease, Krøvel et al. used nanopore sequencing to sequence type Legionella pneumophila3. L. pneumophila can be sequence-typed through characterisation of seven genes, flaA, pilE, asd, mip, mompS, proA and neuA, with at least 3,100 sequence types identified so far3. However, it is challenging to identify L. pneumophila by sequence typing using short-read technology, as the mompS gene is present in two copies in the genome. When the mompS gene copies are non-identical, they may be misassembled or non-typeable using short reads, as short reads cannot cover the entire mompS gene region. The authors sought to overcome these limitations by using nanopore sequencing.
‘This analysis showed that all the isolates that were misassembled or untypeable when using short-read assembly methods were typeable with ONT-only assemblies‘3
Of a collection of 233 L. pneumophila genomes which were sequence-typed with short reads and Sanger sequencing, 35 were identified as being inconclusive due to discrepancies between the technologies; these were taken forward for nanopore sequencing. Libraries were prepared with the Ligation Sequencing Kit and sequenced on a GridION device. Using long nanopore reads, they were able to resolve the mompS region, including in 27 isolates that contained non-identical copies of the mompS gene. The sequence types called with nanopore sequencing were consistent with hybrid assemblies; the authors concluded: ‘we therefore propose ONT sequencing as an alternative method to perform L. pneumophila SBT to overcome the mompS challenge observed with short-reads’.
Avian influenza (AIV) is a highly infectious respiratory disease, which usually infects wild or domestic birds, but with the potential to cause a serious outbreak among humans. Novel genotypes of AIV have previously caused outbreaks in humans, with a lethal variant discovered in Cambodia in 20234. An outbreak of AIV in Europe in 2021–22 was devastating to bird populations, with 40 million birds culled4. Given the economic impact of the disease on agriculture and the potential for a disease epidemic in humans, real-time genomic surveillance is crucial to track circulating stains of AIV. An important facet of this is the ability to rapidly identify links between outbreaks, to determine direct or indirect farm-to-farm contamination routes.
‘ONT technology is highly scalable, from the smallest consumable unit, namely “Flongle”, to high throughput platforms.’3
Croville et al.3 developed an amplicon-based nanopore sequencing workflow for the rapid genetic typing of AIV strains, which they tested on 30 field samples collected from AIV outbreaks in France. Libraries were sequenced in multiplex on a Flongle Flow Cell; real-time alignment was then performed using MinKNOW, which allowed the sequencing run to be stopped as soon as enough data were generated. This method enabled sequential analysis of AIV during outbreaks and the ability to detect minority variants. With a turnaround time of less than 24 hours when sequencing 30 samples, this workflow demonstrates the ability of nanpopore sequencing to provide crucial information for surveillance during an active outbreak.
1. Safiri, S. et al. Front Public Health. 9;10 (2023). DOI: 10.3389/fpubh.2022.1028525.
2. Luo, W. et al. Discov. Med. 35 (176): 332–342 (2023). DOI: 10.24976/Discov.Med.202335176.34
3. Krøvel, A. V. et al. Front Cell Infect Microbiol. (2023). DOI: 20.3389/fcimb.2023.1176182
4. Chang, P. et al. Emerg. microbes & infect. (2023). DOI: 10.1080/22221751.2023.2244091
5. Croville, G. et al. bioRxiv (2023). DOI: 10.1101/2023.05.15.538689






