Blog: Revealing the impact of structural variants on cancer susceptibility
In this blog, Katherine Dixon shares her team’s work on identifying structural variants in cancer genomes, and she explains why identifying them accurately is so important.
 Katherine DixonI am a Ph.D. candidate in Medical Genetics at the University of British Columbia. With a background in cellular and molecular biology, my research focuses on strategies for characterizing the mechanisms underlying cancer predisposition, and improving molecular diagnosis of affected individuals. This includes assessing opportunities and challenges of current and emerging sequencing technologies, as well as the ethical considerations highlighted by challenges in analysis, interpretation, translation and protection of genetic information.
Katherine DixonI am a Ph.D. candidate in Medical Genetics at the University of British Columbia. With a background in cellular and molecular biology, my research focuses on strategies for characterizing the mechanisms underlying cancer predisposition, and improving molecular diagnosis of affected individuals. This includes assessing opportunities and challenges of current and emerging sequencing technologies, as well as the ethical considerations highlighted by challenges in analysis, interpretation, translation and protection of genetic information. Genetic changes that are present in an individual from birth, known as germline variants, contribute to natural phenotypic variability and disease susceptibility. Pathogenic germline variants in cancer predisposition genes underlie around 5-10% of all cancers. Carriers have an increased risk for specific types of cancers, early onset and multiple primary tumours, and sometimes syndrome-related non-cancer phenotypes. Consequently, molecular diagnosis of affected families has important implications for reducing cancer-related morbidity and mortality through increased screening, early detection or preventative clinical interventions.
Recent advances in DNA sequencing technologies, particularly short read-based panel sequencing, have helped uncover the molecular heterogeneity underlying many genetic syndromes. However, the causal germline variants underlying Mendelian and other rare diseases - including cancer predisposition syndromes - are often unknown. Structural variants (SVs) are genomic rearrangements more than 50 bp in length that include both balanced rearrangements, such as inversions and translocations, and unbalanced rearrangements, such as deletions, duplications and insertions (Mills et al. 2011; Chaisson et al. 2019). SVs often occur at regions of the genome with low sequence complexity, repetitive DNA elements, and sequence bias that are inherently difficult to characterize by short-read sequencing. Without an accurate representation of germline SVs, their contribution to cancer predisposition syndromes may not be fully appreciated.
Long-read genome sequencing improves SV interpretation and molecular diagnosis
We recently investigated the utility of nanopore long-read sequencing in resolving the impact of germline SVs in cancer predisposition genes as part of BC Cancer's Personalized OncoGenomics Program, a precision genomics medicine initiative in Vancouver, Canada (Laskin et al. 2015; Pleasance et al. 2020; Thibodeau*, O'Neill*, Dixon* et al. 2020). Using short-read genome sequencing, fourteen of more than 650 individuals with advanced cancers were found to carry germline SVs predicted to impact gene expression, function or splicing. Long-read sequencing resolved multiple germline SVs whose functional impact could not be fully determined through short-read sequencing, including two complex rearrangements with three or more breakpoints. These findings were especially significant for one individual who was found to carry undisrupted copies of each gene on the variant allele; in addition to a lack of phenotype consistent with rare disease, long-read sequencing prevented unnecessary and potentially worrisome clinical genetics referral.
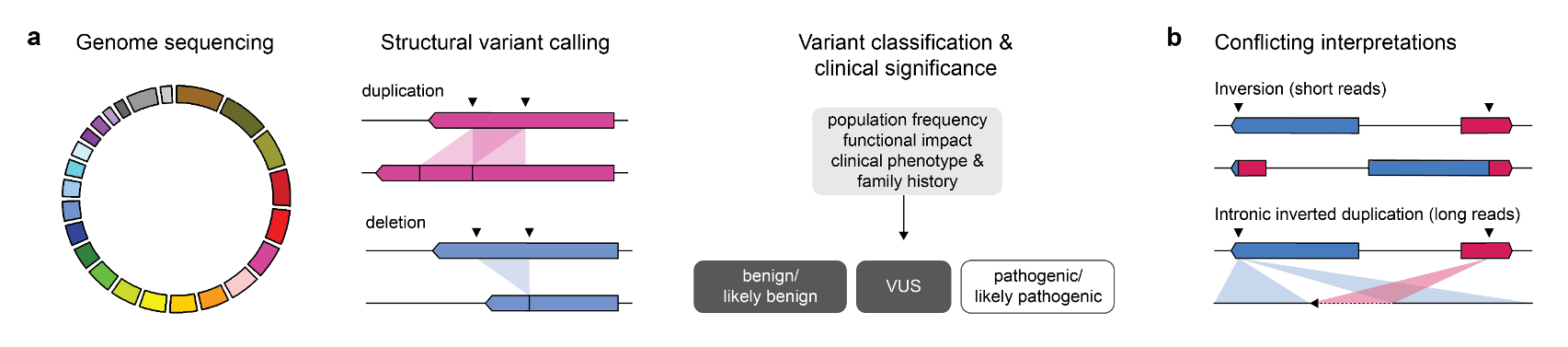
A recurrent variant affecting the tumour suppressor gene TSC2, one of two known causes of tuberous sclerosis complex (TSC), was identified in three individuals. TSC is a highly penetrant autosomal dominant genetic syndrome characterized by benign tumours in multiple parts of the body. Despite convincing short read evidence that a large inversion existed between TSC2 and IFT140, a gene lying more than 500 kb upstream, none of the individuals had a phenotype consistent with TSC. PCR-based assays designed around the predicted breakpoints were unsuccessful in confirming this variant. To our surprise, long-read sequencing revealed that the true variant was a small inverted duplication involving the intronic regions of TSC2 and IFT140. Inaccurate mapping of short reads to homologous sequences near the predicted breakpoints may have resulted in misclassification of this variant as likely pathogenic in the absence of a concerning clinical phenotype.
For several known carriers of pathogenic SVs confirmed by clinical testing, genome sequencing allowed us to precisely map the SV breakpoints. These results are meaningful to inform the development of targeted assays for carrier testing in family members, characterizing recurrent variants and identifying founder variants that are unique to specific populations. Overall, our findings suggest that SVs account for around 10% of pathogenic or likely pathogenic germline variants affecting known cancer predisposition genes. Given the inferential nature of short read-based SV calling, SVs in protein-coding genes and non-coding regions of the genome – which are enriched in repetitive and highly homologous sequences – should not be disregarded as a differential molecular diagnosis in individuals with suspected cancer syndromes.
Integrating long reads into sequencing-based workflows
Long-read sequencing has not only improved our ability to accurately characterize germline SVs underlying rare genetic diseases, but it has more broadly improved our understanding of the complex genetic architecture of the human genome. The clinical validity of sequencing technologies depends on adequate sensitivity and specificity for identifying variants in known disease genes. To address ongoing challenges in germline variant detection, we are developing computational methods for integrating information from short- and long-read sequencing to improve genome-wide SV calling. Further development of targeted nanopore sequencing-based assays is also warranted for the sensitive detection of genetic variants and epigenetic alterations, such as de novo and heritable DNA methylation, at increased coverage and reduced costs (Giesselmann et al. 2019; Gilpatrick et al. 2020). We hope this work will improve our understanding of the molecular basis of rare disease and inform the adoption of novel molecular approaches in the clinic.
Acknowledgments
We gratefully acknowledge the participation of our patients and families, the POG team, the GSC platform, and the generous support of the BC Cancer Foundation and Genome British Columbia (project B20POG). We also acknowledge contributions towards equipment and infrastructure from Genome Canada and Genome BC (projects 202SEQ, 212SEQ, 12002), Canada Foundation for Innovation (projects 20070, 30198, 30981, 33408 and 35444) and the BC Knowledge Development Fund. The results published here are in part based upon data generated by the following projects and obtained from dbGaP: The Cancer Genome Atlas managed by the NCI and NHGRI; Genotype-Tissue Expression (GTEx) Project, supported by the Common Fund of the Office of the Director of the National Institutes of Health.
References
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, Gardner EJ, Rodriguez OL, Guo L, Collins RL, et al. 2019. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun. 10(1).
- Giesselmann P, Brändl B, Raimondeau E, Bowen R, Rohrandt C, Tandon R, Kretzmer H, Assum G, Galonska C, Siebert R, et al. 2019. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat Biotechnol. 37(12):1478–1481.
- Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, Downs B, Sukumar S, Sedlazeck FJ, Timp W. 2020. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol. 38(4):433–438.
- Laskin J, Jones S, Aparicio S, Chia S, Ch’ng C, Deyell R, Eirew P, Fok A, Gelmon K, Ho C, et al. 2015. Lessons learned from the application of whole-genome analysis to the treatment of patients with advanced cancers. Cold Spring Harb Mol case Stud. 1(1):a000570.
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. 2011. Mapping copy number variation by population-scale genome sequencing. Nature. 470(7332):59–65.
- Pleasance E, Titmuss E, Williamson L, Kwan H, Culibrk L, Zhao EY, Dixon K, Fan K, Bowlby R, Jones MR, et al. 2020. Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nat Cancer. 1(4):452–468.
- Thibodeau ML*, O’Neill K*, Dixon K*, Reisle C, Mungall KL, Krzywinski M, Shen Y, Lim HJ, Cheng D, Tse K, et al. 2020. Improved structural variant interpretation for hereditary cancer susceptibility using long-read sequencing. Genet Med.






