The move from reference genomes to pangenomes for advancing genomic equity and personalised medicine
Assembling reference genomes, and the Human Genome Sequencing Consortium (HGSC)
To identify genetic variants, scientists compare an individual's genomic sequence to a standard sequence with defined characteristics, called a reference genome. The first human reference genome was published in 2004, in the journal Nature1, as part of a 13-year research initiative known as The Human Genome Project.
Led by the International Human Genome Sequencing Consortium (HGSC), this project used Sanger sequencing to assemble a genome primarily from the DNA samples of a single donor (13 donors in total). The assembled genome from this ground-breaking effort cost billions of dollars and was just over 90% complete. Limitations with the sequencing technology resulted in numerous gaps, with critical information such as structural variants, notably absent.
The goal of The Human Genome Project was to create an accurate genomic blueprint for the human species, and since 2003, the initiative has continued to pursue this goal. However, a single reference genome cannot accurately represent the enormous diversity of the human species. Instead, we require a wide range of reference genomes, covering diverse backgrounds to build a more reflective pool of genomic data that can be used by clinicians globally.
In 2023, the Human Pangenome Reference Consortium (HPRC) constructed the most complete and accurate human ’pangenome’ ever created2. Drafted from 47 individuals of diverse ethnicities, DNA samples were sequenced and assembled using Oxford Nanopore’s PromethION device, amongst others.
Oxford Nanopore’s technology can directly sequence ultra-long stretches of DNA, including highly repetitive regions, such as telomeres and centromeres, and large structural variants, resulting in comprehensive and contiguous genome assemblies. The high resolution across these challenging regions unlocks the potential to discover disease relevant variants, which are inaccessible with traditional technologies.
The need for pangenomes to illuminate the full spectrum of human diversity
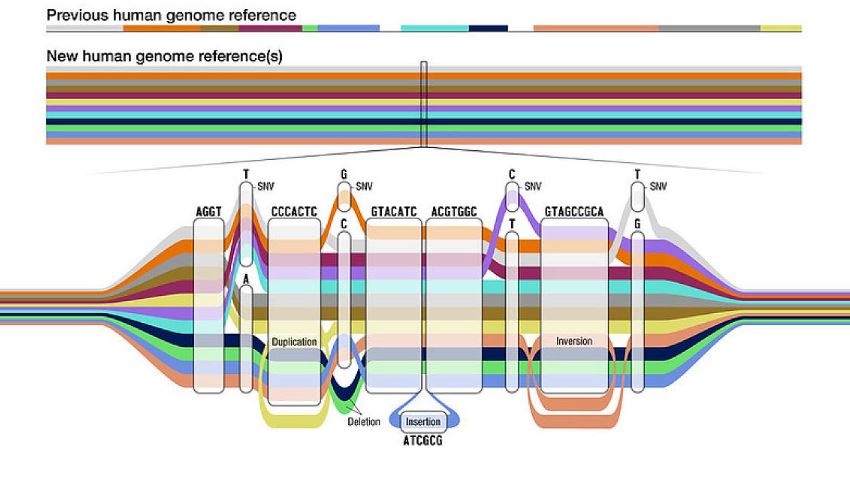
A pangenome represents the complete set of genes within a species or defined population. To build a pangenome, researchers sequence multiple individuals in a population and align their genomes in a design resembling a transient map (Figure 1). Shared sections overlap, and genetic variants branch off, highlighting diversity.
While 99.9% of human DNA is shared between everyone, the remaining 0.1% is responsible for the full spectrum of human diversity. Therefore, having multiple accurate reference pangenomes, which encompass all ethnicities and populations, is critical for accurately comparing genomes, understanding disease risk and development, and potentially informing treatment strategies.
In 2023, the genomic industry saw a surge in novel pangenome assemblies, enabled by nanopore sequencing, especially in underrepresented populations. For example, researchers from the Chinese Pangenome Consortium leveraged nanopore sequencing to investigate 36 ethnic minorities in China and construct a reference pangenome, subsequently published in Nature3. They reported 5.9 million small variants, and 34,223 structural variants that were not reported in the HPRC’s human pangenome draft.
“The missing reference sequences were enriched with archaic-derived alleles and genes that confer essential functions related to keratinisation, response to ultraviolet radiation, DNA repair, immunological responses and lifespan, implying great potential for shedding new light on human evolution and recovering missing heritability in complex disease mapping.”
Furthermore, in late October, first Arab Pangenome Reference (APR) was published4. The APR uncovered 100.93 million base pairs of novel euchromatic sequences absent in previous pangenomes by using nanopore sequencing, among others. This extensive exploration identified 10.68 million population-specific small variants, 108,709 structural variants, and 838 gene duplications, with 13.24% implicated in recessive diseases.
The numerous population-specific variants pointed to the extended period of separation between Middle Eastern populations and other continental groups. Mohammed Uddin and the team expect the future clinical application of this reference to ‘significantly enhance the diagnostic yield for various single gene disorders.’
Lastly, in Australia, the National Centre for Indigenous Genomics (NCIG) utilised nanopore sequencing to construct and publish the first pangenome reference for Aboriginal Australians and Torres Strait Islander communities5. Comprehensive analysis uncovered nearly 160,000 distinct SVs and 137,000 indels, marking the highest numbers observed in any genomic study of isolated communities to date.
“The use of long-read sequencing technology, in combination with the recently completed telomere-to-telomere human reference genome (T2T-chm13), enables us to explore uncharted Aboriginal genomic variation.
Long reads can resolve repetitive or non-unique genes and regions that are intractable with dominant short-read sequencing platforms. Long reads are also superior for the detection of structural variants (SVs), which account for the majority of the differences between the genomes of any 2 individuals and at least 25% of their deleterious alleles, yet are poorly understood owing to technical and analytical limitations.”
Conclusion
To this day, the overarching goal of the Human Genome Project endures: to formulate a complete and accurate genomic blueprint for the human species. By creating several population-based pangenomes, especially in underrepresented communities, researchers are generating a more complete and representative pool of data to help people across the globe. Oxford Nanopore Technologies offers the PromethION range of high-throughput, benchtop devices, capable of sequencing any-read lengths to facilitate these developments, helping to ensure that the benefits of genomic research are applied to all.
-
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). https://doi.org/10.1038/nature03001
-
Liao, WW., Asri, M., Ebler, J. et al. A draft human pangenome reference. Nature 617, 312–324 (2023). https://doi.org/10.1038/s41586-023-05896-x
-
Gao, Y., Yang, X., Chen, H. et al. A pangenome reference of 36 Chinese populations. Nature 619, 112–121 (2023). https://doi.org/10.1038/s41586-023-06173-7
-
Uddin, M., Nassir, N., Almarri, M., et al. A draft Arab pangenome reference, Research Square PREPRINT (2023). https://doi.org/10.21203/rs.3.rs-3490341/v1
-
Reis, A.L.M., Rapadas, M., Hammond, J.M. et al. The landscape of genomic structural variation in Indigenous Australians. Nature 624, 602–610 (2023). https://doi.org/10.1038/s41586-023-06842-7






