Latest Direct RNA Sequencing Kit enables higher accuracy and output
Native RNA sequencing
Direct RNA sequencing is a unique capability of Oxford Nanopore technology. Thanks to the use of nanopores to directly sense information as strands of nucleic acids are fed through, researchers can bypass many of the processing steps that limit — or prevent entirely — the ability of other platforms to detect key details such as base modifications.
Nowhere is this more evident than with RNA. The use of traditional short-read RNA sequencing (RNA-seq) methods limits transcript analysis to ~50–100 base reads, precluding the analysis of full-length transcripts and inhibiting thorough analysis of isoforms. This missing alternative splicing information could provide information crucial to elucidating the molecular mechanisms of disease. Furthermore, the need to convert RNA to cDNA erases RNA modifications, which have known roles in modulating the activity and stability of RNA molecules, as well as in diseases including cancers and neurological disorders.
In contrast, researchers who use nanopore sequencing devices are able to overcome these challenges and obtain a direct look at native RNA molecules. Thanks to the capacity of long nanopore reads to capture long transcripts, direct RNA sequencing delivers isoform-level information for native RNA molecules. It is even possible to directly identify RNA modifications, such as N6-methyladenosine (m6A), which trigger specific and reproducible changes in the signal produced as an RNA molecule passes through a nanopore — negating the need for techniques such as chemical conversion or antibody precipitation. This is effective for many different types of modifications, which can also be distinguished from each other. Libby Snell, Director of RNA and cDNA Sample Technology/Sequencing at Oxford Nanopore Technologies, highlighted how researchers have been ‘using nanopore sequencing technology to learn more about infectious diseases, cancer, epigenetics, and much more’.
Now, the new iteration of the Oxford Nanopore Direct RNA Sequencing Kit (SQK-RNA004) delivers this native RNA information with higher accuracy and increased outputs, allowing even greater exploration of native RNA molecules to provide key insights into translation, transcription, alternative splicing, base modifications, and more. It delivers greater outputs thanks to a faster motor (producing ~30 million reads per PromethION Flow Cell for a human transcriptomic sample) and higher accuracy due to improvements from a new nanopore and enzyme combination, as well as updated architecture for the basecaller. With long nanopore reads, it’s simple to generate full-length transcripts to elucidate gene expression dynamics.
Ultra-rich data
Sample prep is straightforward, taking less than two hours and eliminating many of the steps traditionally used for preparing cDNA-based RNA-seq workflows. Performing a reverse transcription step to create a hybrid DNA-RNA molecule is recommended; however, only the native RNA molecule is sequenced — ‘the DNA strand does not go through the nanopore, but it does help to keep the structure of the RNA molecule stable while it is analysed ’, Libby notes.
Internal benchmarking of the new kit has demonstrated that it allows effective single-read detection of RNA modifications. Shared in a recent poster, this demonstrated that direct RNA nanopore sequencing can call m6A modifications at single-position resolution with high accuracy (Figure 1). The Remora pipeline was used to call these modifications, delivering unprecedented accuracy for a clear view into RNA biology. Validated on a set of five ground truth oligonucleotides containing A or m6A at known positions, the method achieved a median true positive rate of 94.5% and a median true negative rate of 92%.
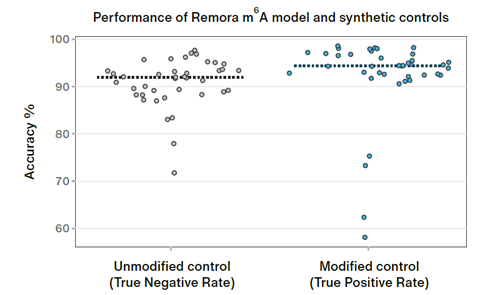 Figure 1. Internal validation of the Remora model demonstrates accurate calling of m6A in native RNA.
Figure 1. Internal validation of the Remora model demonstrates accurate calling of m6A in native RNA.
Prior evaluations have demonstrated that nanopore technology can accurately distinguish between different RNA modifications. The graph below shows how three modifications disrupt signal intensity and other values as RNA moves through the nanopore (Figure 2). In another internal study, direct RNA nanopore sequencing enabled the identification of N1-methyl-pseudouridine (m1Ψ), an RNA modification often found in commercially available mRNA vaccines, with 95% single-read accuracy, demonstrating the potential utility of direct RNA nanopore sequencing for mRNA vaccine quality control workflows.
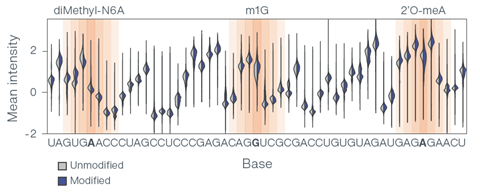 Figure 2. RNA truth sets containing seven modifications (three shown) show significant disruption to the base signal compared to an unmodified control.
Figure 2. RNA truth sets containing seven modifications (three shown) show significant disruption to the base signal compared to an unmodified control.
Direct RNA sequencing also makes it possible to estimate poly-A tail length — a feature which research suggests is an important factor in post-transcriptional regulation — providing further potential to shed new light on gene expression and disease mechanisms. The Direct RNA Sequencing Kit works with the Dorado software tool, which now includes poly-A tail estimation, for a streamlined process, instead of having to turn to a third-party algorithm. In future, this capability will be accessible through MinKNOW — the software onboard nanopore sequencing devices.
Taking all of these features together, Libby describes how ‘in one transcript read, you get the isoform sequence, the 3’ [untranslated region], modifications, and the poly-A tail length — all in one. You don't have to piece it together from different technologies, you don't have to worry about the assembly, because you know what it is. It's all in the same read'.
Learn more about direct RNA sequencing with nanopore technology






