Single nucleotide variants (SNVs) and phasing

Simplify haplotype phasing
Single nucleotide variants (SNVs) have been widely studied for their associations with phenotypic variation and disease; they are also used to phase haplotypes. However, the use of legacy sequencing technology requires PCR, limiting SNV detection to regions amenable to amplification, and short reads make phasing challenging to resolve.
With Oxford Nanopore technology, long, PCR-free sequencing reads are generated, revealing single-nucleotide polymorphisms (SNPs) in regions inaccessible to other technologies, and phasing is greatly simplified.
Featured content

Comprehensive human genomic variant and methylation analysis
From sample to answer, discover how nanopore sequencing delivers comprehensive human variant detection in this best practice, end-to-end workflow. Gain accurate SNV, SV, STR, and methylation results from a single streamlined assay, with integrated tertiary analysis.

Telomere sequencing and phasing
Learn how nanopore sequencing enables precise telomere mapping, length characterisation, and phasing to determine telomere maintenance mechanisms in cancer and telomere shortening with human age.
Related content

The Dark Side of Carrier Screening: Illuminating Hard-to- Decipher Genetic Variation with Nanopore Sequencing

Haplotype-resolved repeat expansions & methylation patterns in 1000 Genome Project data
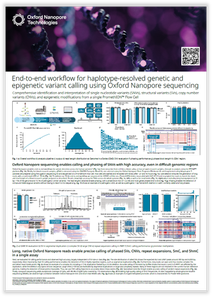
End-to-end workflow for haplotype-resolved genetic and epigenetic variant calling using Oxford Nanopore sequencing
Phasing simplified with Oxford Nanopore sequencing
Recommended device for SNP/SNV calling

PromethION 24
Combining up to 24 independently addressable, high-capacity flow cells with powerful, integrated compute, PromethION 24 delivers flexible, on-demand access to terabases of ultra-rich sequencing data — ideal for comprehensive variant identification across large numbers of samples.






