Assembly
Generate highly contiguous genome assemblies
High-quality genome assemblies are crucial for their use as reliable reference sequences. However, the short reads produced by legacy sequencing technologies lead to highly fragmented, incomplete assemblies. Short reads cannot span important genomic regions, such as repeats and structural variants (SVs), resulting in incorrect assembly. In contrast, Oxford Nanopore technology can deliver long and ultra-long sequencing reads (current record >4 Mb) that can span complex and repeat-rich genomic regions, enabling the generation of highly contiguous reference-quality genome assemblies.
Using Oxford Nanopore sequencing alone, it is now possible to generate complete telomere-to-telomere (T2T) assemblies, including highly accurate Q50 whole human genomes. Register your interest in our T2T product bundle.
Featured content

Capturing global genomic diversity in the human pangenome
Discover how the Human Pangenome Reference Consortium utilised nanopore sequencing in their first draft of the pangenome: a graph-based reference approach representing a broader range of genetic diversity than a single human reference genome.
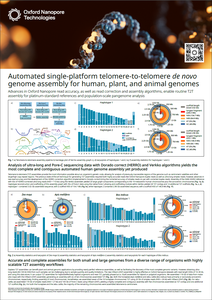
Advances in Oxford Nanopore read accuracy
This poster describes how to achieve accurate and complete assemblies for genomes of any size across a wide variety of organisms, including maize (Zea mays) and honeybee (Apis mellifera) and zebu cattle (Bos indicus), using highly scalable telomere-to-telomere (T2T) assembly workflows.


Recommended device for large genome assembly

PromethION 24
Combine up to 24 independently addressable flow cells with a flexible, large-scale, direct DNA and RNA sequencing device for on-demand access to terabases of data — ideal for high-throughput sequencing and assembly of large and complex genomes.






