London Calling 2021: Day 2
The second day of London Calling 2021 saw more delegates than ever registered, with almost 6000 people signed up to consume the incredible content submitted from all over of the globe. With Plenary, Breakout, Showcase Stage, Mini Theatre, Poster and Spotlight Sessions available, below is a summary of the best parts of the day.
Welcome - Gordon Sanghera
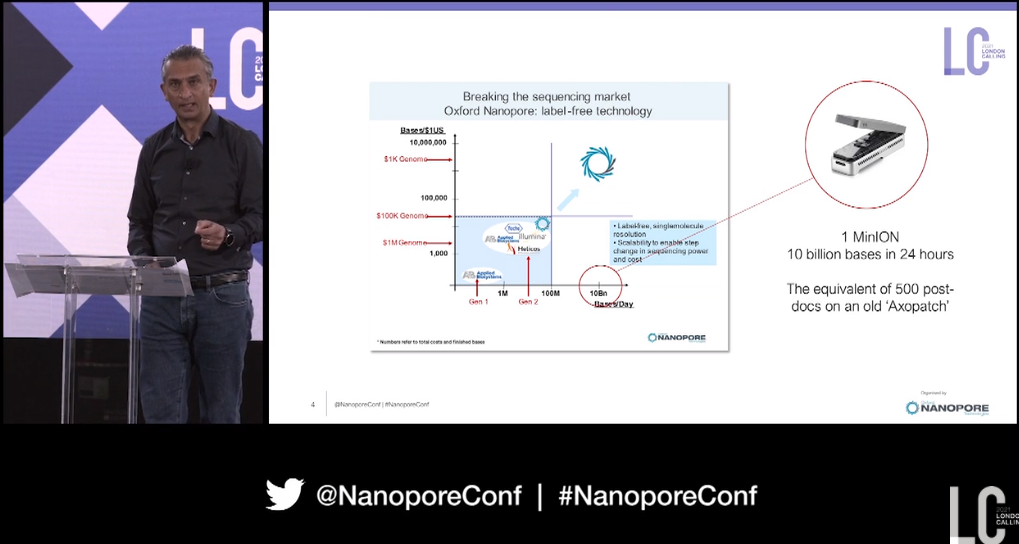
Oxford Nanopore's CEO Gordon Sanghera kicked off the day by referring back to a board slide from 2006, showing how a portable sequencer 'might be useful somewhere' if it was able to produce 10 Gb in 24 hours...'and boy has it been useful'. Round the world we're now seeing decentralized sequencing, with over 250,000 SARS-CoV-2 genomes on GISAID generated from a nanopore device. He highlighted how Oxford Nanopore is committed to global accessibility of our devices, and how this global network will help us all be better prepared for the next pandemic. At the other end of the scale from MinION he turned his attention to PromethION, now capable of sequencing a billion bases per minute. deCODE, Genomics England with the NHS, the American NIH, and the Emirati Genome project were all given as example of how PromethION is now powering population genomics and sequencing at scale with long nanopore reads. With that, he welcomed us to the conference...here we go!
Intraoperative DNA methylation classification of brain tumors impacts neurosurgical strategy - Luna Djirackor
Luna Djirackor covered the astonishing work she and her team at Oslo University Hospital have been conducting with neurosurgical patients during surgery itself. In their department they will see about 300 adults and 30 children with brain tumours every year; commonly, following increasingly worrying symptoms, an MRI scan of the brain will reveal a mass, which ‘you can imagine is completely devastating and so emotionally challenging for the family’. As the tumour type and and subgroups could be highly variable but have an impact on the chosen treatment. it's critical to accurately identify the type of tumour present, as well as its location.
Typically following an MRI scan, surgery would occur within a number of days or weeks to collect a biopsy sample. Histology diagnosis from the sample within an hour or so helps the surgeon to then decide whether to do a partial resection or completely remove the tumour. This isn't what happens every time however — in some instances, after histological analysis, further molecular subtyping can reach a conflicting conclusion. Could they be more specific?
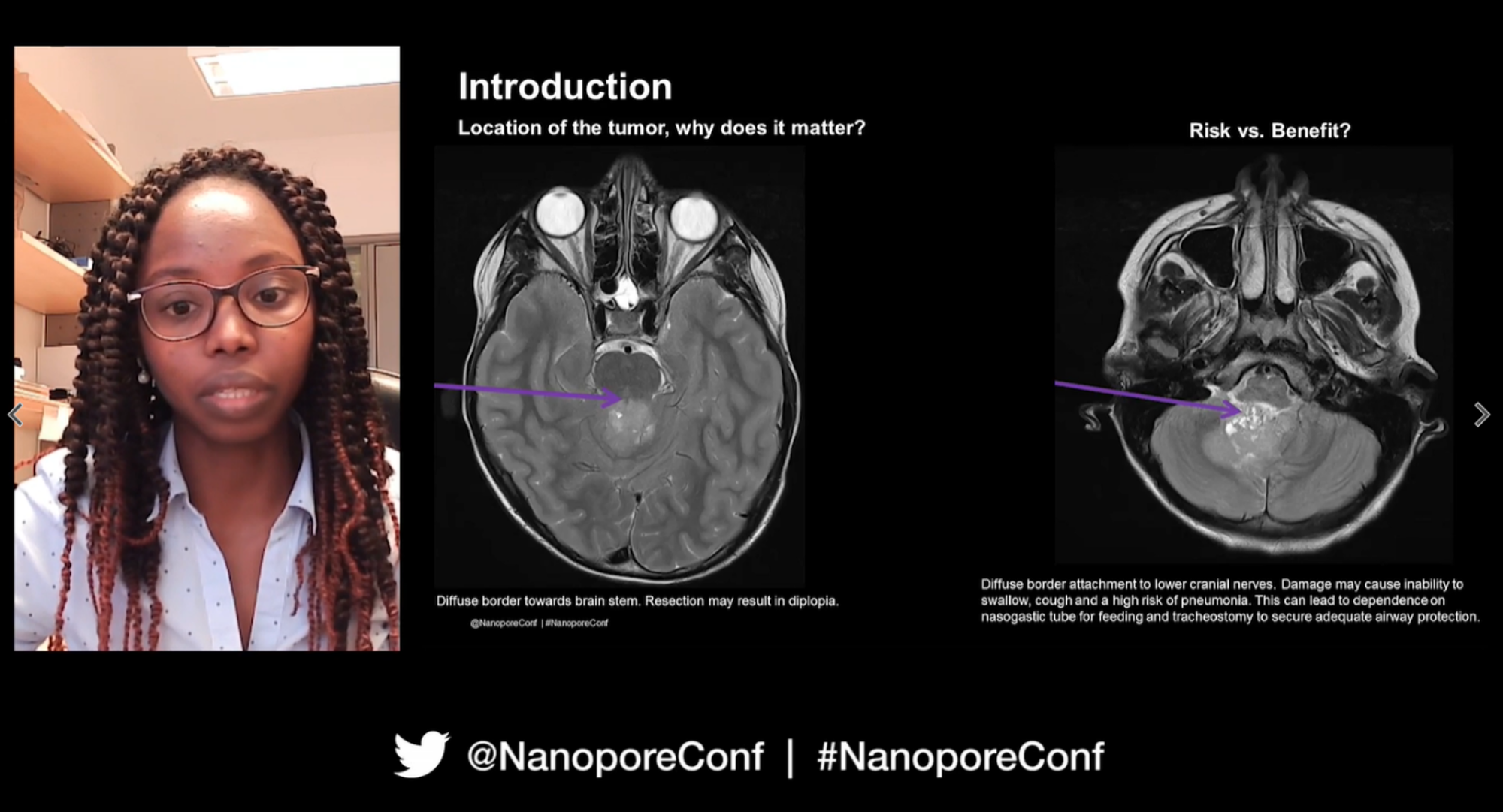
A landmark study from 2018 detailing successful, precise DNA methylation-based classification of central nervous system tumours was impressive, but the turnaround time for analysis was still days to weeks. To make this quicker, Luna and her team took their motivation from Philipp Euskirchen's 2017 talk at London Calling where he presented on how nanopore sequencing showed the potential for same-day, accurate genomic and epigenomic subtyping of brain tumours. For Luna‘that was absolutely amazing’, but she and her team wondered if it could be done even faster. Their approach was to use a nanopore whole-genome sequencing experiment for retrospective and prospective cases, with a methylation classification workflow, using the Rapid Barcoding Kit for library preparation of 10-12 samples per run, and sequencing for 24 hours. One intraoperative run was also performed. The nanoDx pipeline was used for analysis, which contained all the necessary software and outputs a PDF report containing the copy number profile, methylation-based classification results, and a plot of the subtypes.
Their results from research samples of 55 adults and 50 children showed the methylation analysis obtained from a couple of hours of nanopore sequencing matched the pathology results from weeks later. Nanopore DNA methylation analysis also correctly classified tumour tissue that had given inconclusive histology results. This suggested that ‘even on a really tiny little piece of tissue with not too much tumour’ the nanopore sequence data could get the correct result. Taking this further, they then tested an additional 20 intraoperative research biopsy samples. The first report could be returned within 91-161 minutes. Their sequencing statistics suggested that only 30 minutes, or sometimes less, was required to detect ~3,500 CpG sites, which usually provided ‘a really solid call’. Out of the 20 cases studied in this research project, the data suggested that nanopore DNA methylation analysis had the potential to impact surgical strategy and management in 12 of those cases.
Luna closed by extending her gratitude to those patients and parents who took part in the study, to her team, to Philipp Euskirchen’s team in Berlin, and to her funders.
Detection of differential isoform expression and usage during cellular differentiation using long read RNA sequencing - Wilfried Haerty
With work from the Earlham Institute, UK and in collaboration with researchers from Oxford, Wilfried Haerty covered how long read nanopore RNA sequencing can elucidate differential isoform expression within neuronal cells. Alternative splicing of transcripts is a highly regulated process, but aberrant splicing is known to be associated with several diseases, such as early-onset Parkinson’s disease, and it's also a hallmark of several cancers. Therefore it's important to identify alternatively spliced transcripts.
He covered how short-read RNA sequencing can be of limited benefit for these investigations, partly due to the need to computationally assemble short reads into inferred potential transcripts; the literature suggests that ‘up to 50%’ of transcripts identified via short-read sequencing are not correctly assembled. This leads to ‘a very confusing picture’ when trying to assess splicing at gene level. A human example is the CACNA1C gene, which is a major candidate risk gene in neuropsychiatric and cardiovascular disorders. It comprises over 50 exons, and using nanopore sequencing, they have identified 241 novel transcripts above those already annotated (Clark et al. Mol. Psychiatry. 2020).
Wilfried’s research, along with the Oxford team, involved investigating splicing regulation during neuronal cell differentiation. Of the genes found not to be differentially expressed, 1,276 were found to have at least one differentially expressed transcript. ‘This was very surprising’ but also ‘highlighted the fact that we really, really need to go to the transcript level to be able to capture things that we would never have been able to do if we were staying at the gene level’
Wilfried wrapped up by stating that long nanopore reads can be used to quantify gene and transcript expression, ‘with very good sensitivity’. Crucially, the identification of differential transcript expression without differential gene expression provides evidence for differential transcript usage with potential functional consequences.
You can read the pre-print from Wilfried and his team on bioRxiv: Wright et al. 2021. DOI: https://doi.org/10.1101/2021.04.27.441628
Accurate variant calling and de novo assembly with nanopore reads - Kishwar Shafin
Moving from the transcriptome and back to the genome, Kishwar Shafin (University of California, Santa Cruz, USA) outlined that his talk would focus on two key topics: the haplotype-aware small variant calling pipeline PEPPER-Margin-DeepVariant, and updates to the Shasta de novo genome assembler.
The precisionFDA truth challenge v2 in 2020, which aimed to assess and benchmark variant calling in difficult-to-map regions across three different sequencing platforms, awarded Kishwar and his team with the winning variant calling performance in both the benchmark regions and difficult-to-map regions. Kishwar explained that the longer reads produced by nanopore sequencing enable reads to be aligned with higher confidence. ‘This is the first time we have seen Oxford Nanopore outperform short-read based variant calling in identifying single nucleotide polymorphisms’.
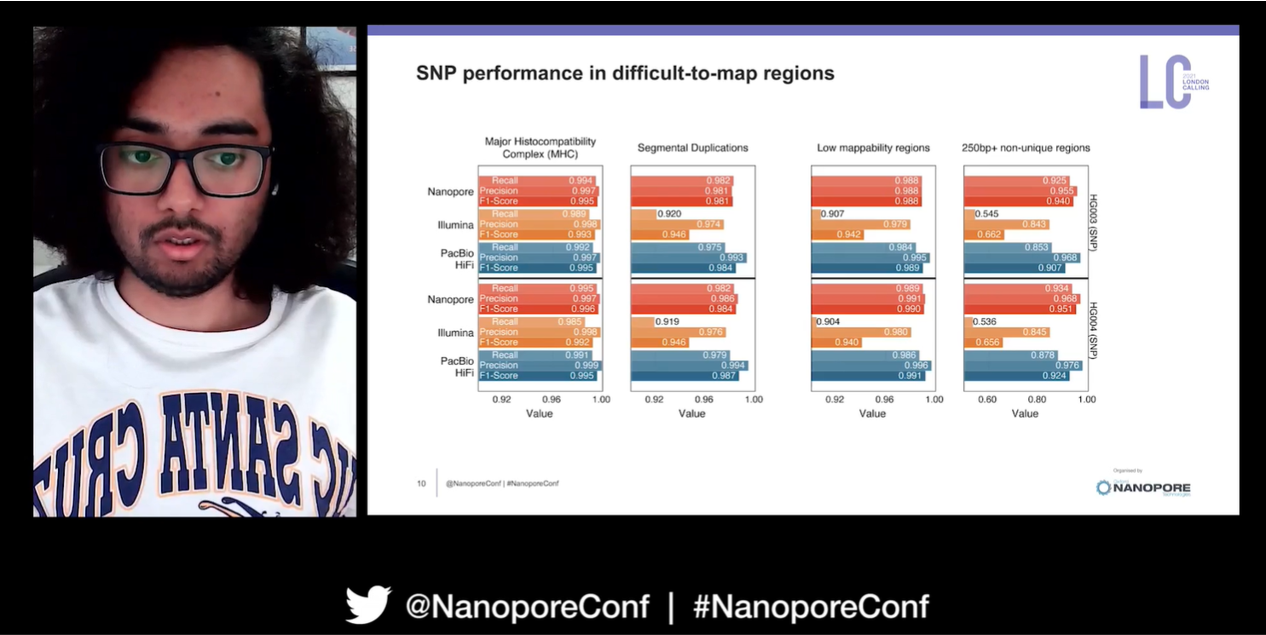
They are not yet done with improving their methods, and are looking into ALT alignment in DeepVariant to improve variant genotyping of the alternative allele, as well as stratified variant calls. They have been working with the very latest data from Oxford Nanopore's R10.3 Q20 chemistry also, a data type which ‘produces extremely high-quality reads’. Training PEPPER-DeepVariant with this data (R10.3 Q20, Bonito, 60X HG002 Chr20), they found significant improvement in SNP identification and INDEL calling.
Not content with a best-in-class variant detection pipeline, Kishwar then focused on his team’s updates to the Shasta de novo assembler; in particular, iterative assembly was found to significantly improve contiguity in the CHM13 human genome assembly, increasing NG50 from 65.3 Mb to 90.6 Mb.
To close, he discussed tackling the problem of centromere assembly with high identity, as well as using PEPPER-Margin-DeepVariant pipeline to polish de novo assemblies in a diploid manner. Future work will involve incorporating the Flye polisher, in collaboration with Mikhail Kolmogorov, into their PEPPER-Margin pipeline, with a further extension to integrate Guppy methylation calls into their small variant workflow.
Structural variant detection from long-read sequencing data with cuteSV - Tao Jiang
Structural Variants (SVs) range from 50 bp to several megabases in length and can be classified into 3 categories; inversions and translocations; deletions, insertions, and duplications; and complex rearrangements. SVs have been detected throughout the human genome, with noticeable effects associated with complex phenotypes and diseases. Due to their impact, there is high value in comprehensive detection and understanding of SVs and their evolution. Here Tao Jiang of of Harbin Institute of Technology, China, took us through the possibilities for SV detection using cuteSV.
Long sequencing reads from Oxford Nanopore technology enable ‘high resolution’ and long-range SV analysis. Tao noted that nanopore sequencing technology provides benefits including ultra-long reads, direct sequencing, and lack of GC bias. These benefits aid SV detection, particularly in repetitive regions. Challenges do remain in SV calling, including the demand for high coverage to resolve complex variants, unsuitable clustering rules for SV prediction, and high compute requirements for some analyses, which may not be compatible with small computing facilities. To try and address these bottlenecks, Tao has developed cuteSV.

Tao noted three typical applications of cuteSV: long-read alignment-based SV calling; discovery of SVs using diploid-assembly alignments; and SV calling on a large scale in populations. He demonstrated high precision and recall of SV calling and genotyping using cuteSV in 47X HG002 PromethION sequencing data. The cuteSV workflow improved SV detection compared to two other ‘state-of-the-art’ SV callers, SVIM and Sniffles. Discussing these results further, Tao demonstrated that the workflow uncovered a higher number of insertions than deletions in low-complexity regions and highlighted how the results could be associated with known diseases such as psoriasis. He impressively compared cuteSV to alternative tools, summarising it's ability as a ‘new powerful method that allows the pairwise comparison of genomes and enables SV calling even in the absence of a suitable reference genome’. This was further shown by its impressive performance on examples where only 15X and 30X of HG002 PromethION sequencing data was used.
Typically, Tao states three applications of cuteSV. Firstly, long-read alignment-based SV calling, based on read mapping with tools such as minimap2, LRA, and NGMLR. Secondly, the discovery of haplotype-specific SVs using diploid assembly alignments. And thirdly, cuteSV also enables SV calling in large-scale populations. Despite these existing use cases he intends to work on further improvements to the SV calling pipeline, including production of consensus sequences of each alternative allele, and further improvements of accuracy of breakpoint detection and size. A second aim is to develop a new joint SV calling method to improve SV calling performance at cohort level.
From flu to you: sequencing at any scale - Dan Turner
With a witty talk title and a spring in his step, Oxford Nanopore's Dan Turner took us on a journey of sequencing from viruses to the human genome. Over the past 18 months the Oxford Nanopore Apps team has been using nanopore sequencing technology to help mitigate the spread of and understand the genetics of the SARS-CoV-2 virus. This has included support and development of well-known virus sequencing protocols, such as the SISPA protocol for metagenomic analysis of respiratory samples, and the ARTIC method for SARS-CoV-2 genome sequencing. Spike-seq is also being used for for targeted, highly-multiplexed analysis of the SARS-CoV-2 spike gene to identify e.g. variants of concern. An adaptive sampling application relating to this work came in collaboration with the researchers at the Mount Sinai School of Medicine, with whom the team have been investigating host responses to COVID-19 vaccination focusing on the IGH locus combined with single-cell transcriptome analysis of B-cells (see the Applications poster for more information).
Going ‘beyond the COVID pandemic’, Dan described how they have been looking to ‘get a head start’ on what the media have warned could be a severe flu season this winter. His team have been developing whole-genome sequencing protocols able to precisely identify influenza type, sub-type, and genomic variants to assist genomic epidemiology efforts and vaccine selection. This utilised a novel library prep method facilitated by the vaccinia virus topoisomerase enzyme, combining the advantages of both rapid and ligation-based preparation to produce a method that is fast, has good control over fragment length, and is easy to automate.
This approach could be applied not just to influenza genome sequencing, but also as a general library preparation method for whole-genome sequencing, as it is simple, quick, gives good control over fragment length, and should eliminate any chimeric reads. Dan explored how 'topo' library prep could be ideal for sequencing ultra-long reads, potentially making a 4.4 Mb read seem ‘lacklustre’ in the not-too-distant future.

Dan next covered developments that have been made in constructing haplotype-resolved assemblies. ‘In the ideal world’ we want haplotype-resolved assemblies, with chromosome-length scaffolds consisting of single haplotypes. ‘Using long reads are a very good place to start’ here; Dan showed graphs demonstrating how longer reads give rise to longer phase blocks, with a 30X ultra-long dataset producing a phase block length of 12.61 Mb for chromosome 21.
Another approach is to use orthologous methods, such as chromatin conformation capture methods that uncover the three-dimensional organisation of the genome, e.g. Pore-C. Individual long Pore-C reads contain different segments from the same homologous chromosome, so can be used to help resolve haplotypes. Dan displayed graphs demonstrating increased haploid human genome contiguity with the addition of Pore-C data: from 45.3 Mb N50 to 152.2 N50. ‘The longest scaffold that we’ve made is…over 230 megabases…which corresponds to… the entire chromosome 2’. As far as he was aware ‘this is the most contiguous haplotype-resolved human assembly to date’. He emphasised that all the information presented here comes from just two PromethION Flow Cells, one to produce the ultra-long reads, and one for the Pore-C data. This represents a ‘great leap forward’ when it comes to population-scale generation of haplotype-resolved assemblies.
If this sounds like something for you then please register your interest here for the haplotype-resolved assembly workflow: https://register.nanoporetech.com/haplotype-resolved-assembly-workflow
Genetic and epigenetic profiling of complex chromosomal rearrangements in a Sonic Hedgehog medulloblastoma sample from a patient with Li-Fraumeni Syndrome - Rene Snajder
Li-Fraumeni syndrome is underpinned by mutations in the gene encoding for the tumour supression protein TP53. This syndrome often culminates in patients experiencing multiple cancers throughout a lifetime, so with that knowledge the benefit of the work being done by Rene Snajder of the German Cancer Research Centre is clear for all to see. He wants to disentangle the effects of structural variations and epigenetic changes that drive formation of medulloblastoma brain tumours, specifically the Sonic Hedgehog Medulloblastoma (SHH-MB); these are typically associated with large-scale chromosomal rearrangements. Armed with multiple data sets available from a single patient he had everything required to delve into one specific example.
Copy number profiles of the patient's genome showed some striking aberrations, including a loss of heterozygosity in chromosome 3 and a drastically rearranged chromosome 7. Using long nanopore reads they then called large structural variations from the primary tumour data. He found a significant number of intra and inter-chromosomal rearrangements, and the unexpected discovery of rearrangements between different chromosomes. When zooming in on these inter-chromosomal rearrangements, there appeared to be two main clusters - the most obvious was between chromosome 11 and chromosome 17 which appeared to have created a novel chromosome. Using nanopore long-read data, they succeeded in assembling 1.7Mb of the novel chromosome, containing 57 genes in total, of which 22 were protein coding. Rene and his team further validated their hypothesis of a novel chromosome by using FISH, and found signals of chromosome 11 and 17 in much closer proximity in the medulloblastoma tissue when compared to the healthy donor tissue.
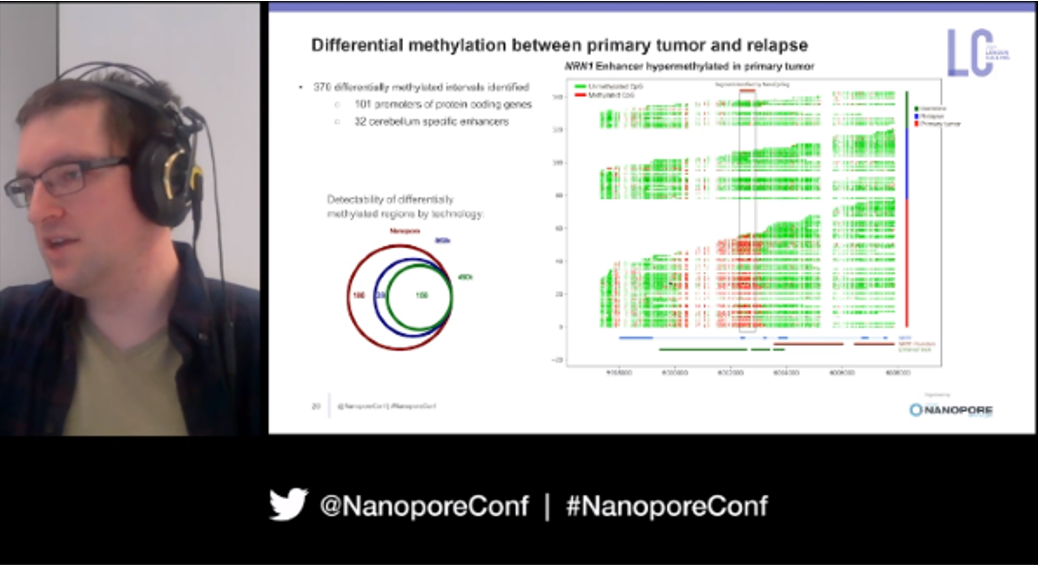
Not content solely with information on structural changes, next Rene moved to the epigenetic facets of his experiments. Using Nanopolish and raw nanopore signals for 5-methylcytosine at CpG sites on the primary tumour and relapse sample, he discovered 370 differentially methylated regions, 101 of which overlapped with promoters of various protein coding genes and 32 were cerebellum-specific enhancers. In the process of this he created his own MetH5 File Format, which efficiently stores and retrieves methylation predictions without losing the read-level information provided by nanopore sequencing; 'you can never have too many file formats' were his sage words.
Following this he briefly noted his findings on validating gene fusions using SVs called from long reads (they could disentangle how the gene fusions may have arisen), and concluded his work succinctly; they were able to further understanding of SHH-MBs through chromosome arm phasing, constructing targeted assemblies to resolve SVs, and use nanopore sequencing for comprehensive methylation profiling and understanding complex gene fusions. Amazing work by Rene and his team.
Advancing long-read genome sequencing towards improved clinical and molecular diagnosis of rare genetic disease - Katherine Dixon
Our final plenary for Day 1 came from Katie Dixon of Canada’s Michael Smith Genome Sciences Centre. With statistics mirroring those presented in the Showcase Stage rare disease investigations, she explained that a rare disease is defined as a specific disease occurring in fewer than 1 in 2,000 individuals (UK) or 200,000 individuals (US); worldwide, 260-450 million people have a rare disease, so ‘collectively they pose a significant burden on the health system’. Many rare diseases (~70-80%) are suspected to be genetic in origin. To show the progressive thinking required for rare disease investigations, Katie shared that there is a saying in medicine from Theodore Woodward: ‘When you hear hoofbeats, think horses, not zebras’; however, in rare diseases, you have to think of zebras.
Diagnosis of rare disease faces many challenges; long duration of a "diagnostic odyssey", genetic and phenotypic heterogeneity, altered clinical phenotypes despite similar genetic abnormalities, and unresolved complex variation. This last one in particular suffers from the he technologies most widely used for genetic diagnosis, such as short-read sequencing. Katie and her team investigated the use of nanopore sequencing to identify complex variants such as these in clinical research samples, with a publication from Thibodeau, O’Neill, Dixon et al. in Genetics in Medicine 2020 elaborating on how long nanopore reads ultimately helped explain the lack of a particular phenotype observed in the patient from which the sample had been sourced.
This was extended in additional work on 32 individuals sequenced to 30x coverage depth to identify SVs that may not have been identified with standard testing methods. She suggested ‘that nanopore genome sequencing at 30X could potentially be a replacement for other short-read based genome sequencing in the future’. Additional insights into SVs known for breast cancer-predisposition genes were also carried out on the cohort. Nanopore sequencing was also able to detect founder deletion variants as well as duplication events; Katie suggested that ‘targeted long-read sequencing may be a good alternative, and a cost-effective alternative, to approaches like targeted Sanger sequencing or MPLA for copy number analysis’.
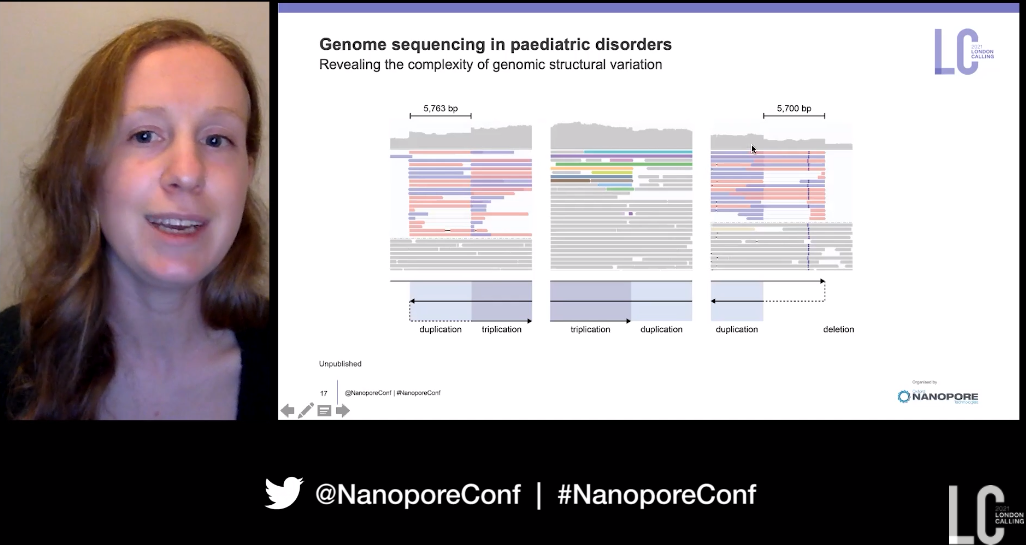
Switching focus to paediatric disorders, cases here tend to undergo a wider variety of tests than adult rare disease. Short-read whole-genome sequencing has only ‘marginally improved’ the rates of diagnosis from exome sequencing, which may be due to limitations in its ability to identify certain types of genetic variation. In one example of a child with anopthalmia, microcephaly, hypertonia, hepatosplenomegaly, and seizures, a chromosomal rearrangement had been identified involving duplication, triplication, and deletion in chromosome 13q, using standard testing. However nanopore sequencing was essential in this example to reveal the underlying complex SVs, revealing a specific signature of inverted sequences, and resolving the order of the complex rearrangement, involving duplications, inverted triplication, and a terminal deletion.
Katie signed off by indicating her team aim to uncover dark regions of the genome with nanopore sequencing, which are difficult to access using other sequencing technologies. In future, they aim to investigate how targeted sequencing could be used for rapid, sensitive, and cost-effective analysis of well-characterised candidate disease genes.
Oxford Nanopore tech updates
Please find a summary and recordings of the key tech updates here






