Interview: Using PromethION for Cancer, Collaborations and more

with Dr Richard Moore, Sequencing Group Leader at the Genome Science Centre, BC Cancer
Conducted and written by Jonathan Pugh
"We are routinely getting 100-120 gigabases out, that’s standard...often we’re getting 140, 150”
Dr Richard Moore on data yield from individual flow cells on their PromethION 24, a device capable of running up to 24 flow cells
Despite its name, the Genome Science Centre at BC Cancer tackles a wide variety of sequencing challenges. In my discussion with Sequencing Group Leader Dr Richard Moore we touched on the variable operations run by his team, from de novo sequencing “lots of critters” to RNA sequencing and rapid turnaround of Neonatal Intensive Care Unit (NICU) samples. Any given day they may have “10-15 projects…100 or so collaborators doing something at the same time”, but 80% of their work is still cancer-focused. For their PromethION-based nanopore sequencing (for which they use a PromethION 24 which can run up to 24 individual flow cells) they have three main interests; de novo assembly; cancer studies; and full-length RNA isoform sequencing.
Flexible device
Running such varied operations on a daily basis requires careful thought. “We’ll make 4 libraries a day, but the sequencer [PromethION] might have 12 running at once… some have more demand on a turnaround, like these rapid turnarounds for the sick children in [the] NICU, so we try to get them on as soon as they arrive. Otherwise it’s first in first out. So if it’s RNA libraries that are coming in, cDNA, then they’ll be done in parallel with the genomes”. If surge capacity is required as vital samples arrive this is no problem for the PromethION, with its modular nature offering 24 or 48 positions.
Multiplexing also provides additional flexibility and is their favored approach during technical development on their de novo samples; “Now when we’re trying to validate a protocol we want to go with we tend to run it straight on the PromethION. Especially with barcoding we can then try lots of things at once… it can be a little tricky when you try and compare yields that way, but you can certainly look for the distribution of sizes etc and whether something’s working or not quite accurately”.
Optimising output
The team pay close attention to their experimental results to ensure things perform as expected. For their genomic samples “size distribution is a big one. And then quality, you’re looking at how well it’s performing yield wise, so number of reads as well as Gigabases”. Understanding what a good sequencing run looks like gives them confidence in achieving consistent results. “When we get a high quality sample in…say we get a blood sample in and we do our extraction on it, then it’s very consistent”. They can see more variation in their sequencing when things haven’t been done consistently, “especially cancer samples that have been stored in different ways, frozen in different ways, extracted in different ways”.
"More often than not we’re going all the way through to taking the lead on writing a publication…for things like narwhal, dolphin, orca, things like that, grizzly bear we’ve written those papers"
Their desired sample preparation method uses MagAttract for high molecular weight gDNA extraction, followed by G-tube shearing for fragmentation; “for those ones we are routinely getting 100-120 Gigabases out, that’s standard…often we’re getting 140, 150 [Gigabases]”. Older samples which have been stored may have fragmentation and are “slightly sheared already so the size distribution profile is less tight, but you get a longer tail…depending on the projects…those long tails are very interesting, those 100 kb reads or more”. Their cancer studies tend to have read N50s in the 15-20kb range.
A focus on cancer
 The expertise of the Genome Science Centre team is widespread, but predominantly they work on cancer studies and are discovering some fascinating new biology aided by characteristics of nanopore sequencing — such as long reads and native DNA base modification. Vanessa Porter of Dr Marco Marra’s lab, who gave an update on her project at London Calling 2021, is identifying genomic and epigenomic factors associated with human and viral gene dysregulation related to human papillomavirus (HPV) integration events – when the HPV genome inserts into the human genome. From her work, phasing and read-specific methylation calling suggests integrated virus contributes to demethylation of regulatory regions near cancer genes, having a direct involvement with cervical cancer development.
The expertise of the Genome Science Centre team is widespread, but predominantly they work on cancer studies and are discovering some fascinating new biology aided by characteristics of nanopore sequencing — such as long reads and native DNA base modification. Vanessa Porter of Dr Marco Marra’s lab, who gave an update on her project at London Calling 2021, is identifying genomic and epigenomic factors associated with human and viral gene dysregulation related to human papillomavirus (HPV) integration events – when the HPV genome inserts into the human genome. From her work, phasing and read-specific methylation calling suggests integrated virus contributes to demethylation of regulatory regions near cancer genes, having a direct involvement with cervical cancer development.
“all of our human genome runs, we’re running the methylation run too”
In addition to whole genome sequencing (WGS), Vanessa has used the PromethION for whole-transcriptome sequencing of HPV tumours, from which she has identified a highly expressed transcript from an HPV integration which to the best of her knowledge has never been described before. Transcriptomics work extends beyond Vanessa’s project as well, with a number of leukaemia collaborators interested in alternative splice variants which nanopore sequencing is very much suited to, thanks to its ability to read full-length transcripts and shed more light on the varied nature of transcript isoforms and splicing.
Structural variants
Also illuminating new insights into genomic conditions is Vanessa’s colleague and BC Cancer team member Katherine Dixon who spoke at London Calling on improving rare genetic disease diagnosis using nanopore sequencing. An estimated 260-450 million people world-wide have a rare disease, with ~70-80% expected to be genetic in origin. 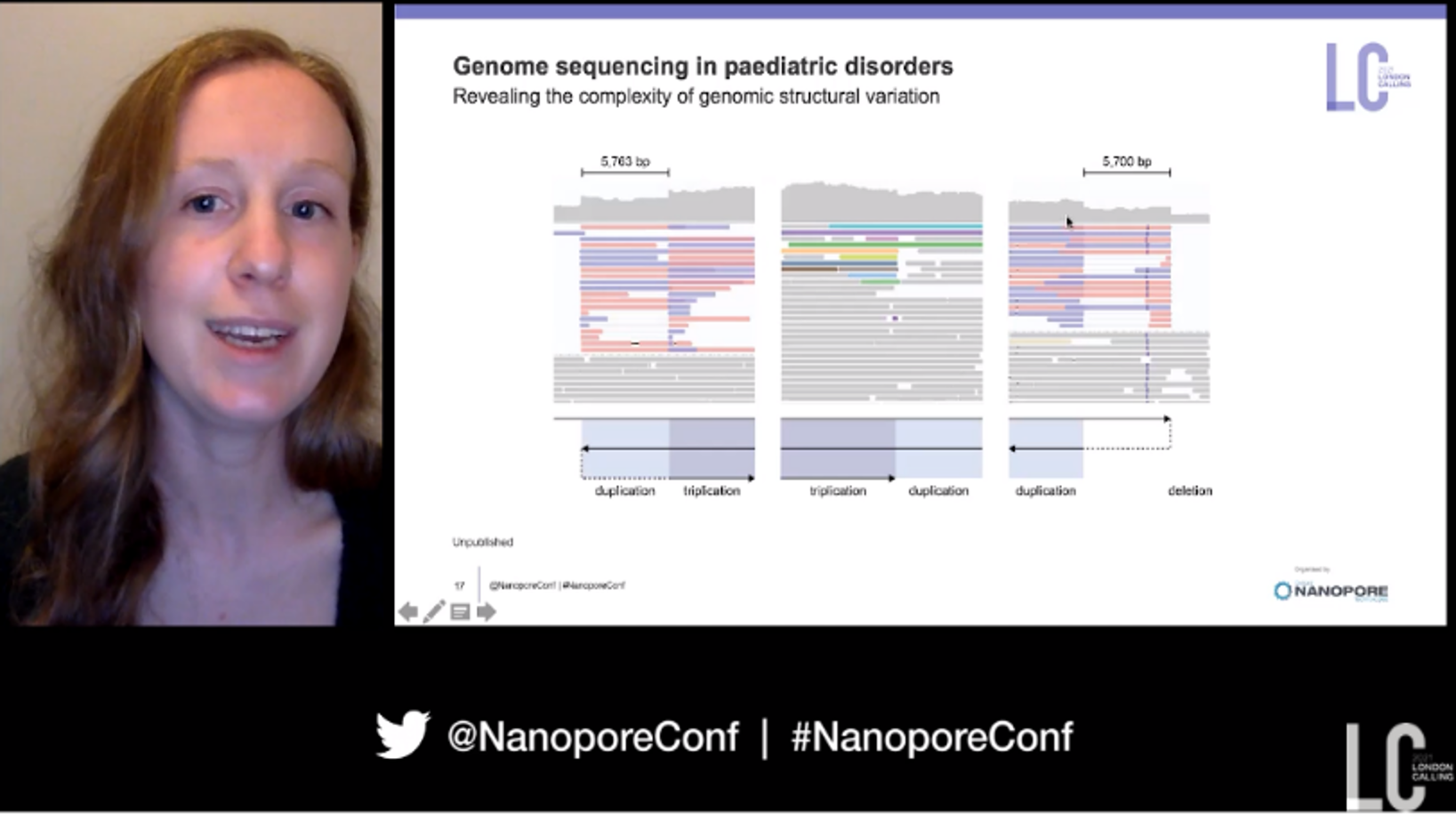 Structural differences or ’structural variants’ in their genomes are one aspect that should be considered in rare disease diagnosis, but in Richard’s words they have “been finding and resolving a lot of structural variation that’s been difficult to resolve on the short read platforms”. Using the PromethION Katherine has performed WGS on a number of individuals, including children, and the resultant complexity of structural changes and chromosomal rearrangements observed has led her to suggest that “nanopore genome sequencing at 30X could potentially be a replacement for other short-read based genome sequencing in the future”.
Structural differences or ’structural variants’ in their genomes are one aspect that should be considered in rare disease diagnosis, but in Richard’s words they have “been finding and resolving a lot of structural variation that’s been difficult to resolve on the short read platforms”. Using the PromethION Katherine has performed WGS on a number of individuals, including children, and the resultant complexity of structural changes and chromosomal rearrangements observed has led her to suggest that “nanopore genome sequencing at 30X could potentially be a replacement for other short-read based genome sequencing in the future”.
Methylation and de novo assembly
It’s clear Richard and his colleagues are getting the most out of their PromethION 24. Asides from long reads, high single-sample coverage, rapid sequencing and full-length transcripts, they also take advantage of “being able to do the methylome directly”, an area already touched on with Vanessa’s work and further referenced by Richard almost casually: “We’ve also been looking for methylation on it [the PromethION], seems to work quite well actually”. Their work was published earlier this year in Genome Biology with the introduction of two new tools; 1. NanoMethPhase (phasing 5-methylcytosine calls from nanopore sequencing), and 2. SNVoter (nanopore SNV post-processing). These tools ‘can accurately detect allele-specific methylation genome-wide using nanopore sequence data with low coverage of about ten-fold redundancy’. With these tools in tow now for “all of our human genome runs we’re running the methylation” too.
Their final large project area is de novo assembly, for which they throw the net wide and work with many collaborators on a range of species “from algae all the way up to mammalian genomes”. Their involvement came through the CanSeq150 programme, which aimed to achieve reference-assembly quality genomes of 150 new animals as chosen by the research community. “More often than not, we’re going all the way through to taking the lead on writing a publication…for things like narwhal, dolphin, orca, things like that, grizzly bear, we’ve written those papers”. Even here his team can’t help but dive deeper to see what they can find, playing detective for some aquarium samples and attempting to determine the cause of death. Slightly disappointed, he informs me there was “no smoking gun in the ones we tried, but it’s an interesting thing to do”.
After my call with Richard has finished and I’m reading back through my notes, I can’t help but feel slightly in awe of the quantity and quality of work he and his team get through on the PromethION. Never in one call before have I been given insights ranging from cervical cancer cohorts in sub-Saharan Africa to the similarities between polar and grizzly bear, or how NICU samples may be demonstrating the value of investigating unique methylomes in sick children. The amazing work conducted at BC Cancer is reflected in their literary library and the quality of their conference talks, and I am genuinely excited to see what this talented and forward-thinking group deliver as their use of nanopore sequencing continues.
Jonathan Pugh is an Associate Director at Oxford Nanopore Technologies and has spent almost 10 years developing and introducing nanopore sequencing technology
Want to learn more?
Read more about the BC Cancer Genome Sciences Centre
Get more information on Structural Variants and nanopore sequencing






